Overview¶
Capture Oligo Design¶
oligo provides functionality to automate primer design for DNA capture experiments, providing the user with details about efficiency of the primers generated.
Installation¶
Local¶
oligo¶
To install oligo on your local machine, it is recommended to first create a new Python environment ( >=3.8 ) using your preferred method e.g. conda, pyenv etc. Once the new environment is activated, install oligo
via pip. Note that the package is called oligo but has a published name of oligo-capture to ensure it had a unique name in the public repository.
$ pip install oligo-capture
Ensure it has installed correctly by running the following command and verifying that you see the installed version in the standard output
$ python -m oligo --version
oligo v0.2
Before running the full oligo pipeline you will need to install RepeatMasker and either BLAT or STAR, depending on how you intend to run oligo (see below for more details)
Dependencies¶
Since running oligo locally requires a number of third-party software to be installed, users might find it less hassle to run
oligovia the Docker image, if this option is available to them. See the Docker section for more details.
- RepeatMasker
oligouses RepeatMasker (RM) to determine if oligos contain simple sequence repeats as these can reduce the efficiency of the oligo for targeted capture. Follow the instructions on RepeatMasker home page for installing RM on your local system. The most recent version of RM thatoligohas been tested with is v4.1.5. As detailed on the RM home page, its installation depends on a Sequence Search Engine (it is recommended to use HMMER foroligo) and Tandem Repeat Finder (TRF). For the Repeat Database, RM ships with the curated set of Dfam 3.7 Transposable Elements which is sufficient but users are free to use the full set if required; further instructions are on the RM home page.
The RM home page mentions that it requires the Python library
h5py, however this is listed as a dependency of theoligopackage so will already be installed in your Python environment from when you ran thepip installstep.
- BLAT
oligouses the BLAST-Like Alignment Tool (BLAT) to determine any off-target binding sites of an oligo within the genome, in addition to its intended binding site. An oligo that binds to multiple regions will have a reduced score since it will perform a less-specific capture. BLAT executables can be found by going to http://hgdownload.soe.ucsc.edu/admin/exe/ and locating the BLAT directory in the for your systems archtecture. For example, for Linux.x86 architecture,rsyncshould be used to get the BLAT executables on your system:
$ rsync -aP rsync://hgdownload.soe.ucsc.edu/genome/admin/exe/linux.x86_64/blat/ ./
See http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/blat/ for more details.
Please note, that as specified on its website, “the Blat source and executables are freely available for academic, nonprofit and personal use. Commercial licensing information is available on the Kent Informatics website (http://www.kentinformatics.com/)”. Ensure that you are adhering to this licence agreement if you are using
oligowith--blatenabled.
- STAR
As an alternative to BLAT,
oligoallows users to use the Spliced Transcripts Alignment to a Reference (STAR) alignment program for increased speed when determining multiple binding events for oligo sequences. BLAT is more widely used to detect off-target binding events, however BLAT can be particulary slow for large designs, especially for the human reference genomes. STAR’s exceptional speed is better suited for designs with >1000 oligos. If you think you would prefer to use STAR instead, visit the STAR GitHub page for instructions on how to install it.
Docker¶
Due to oligo requiring various third-party software, it can instead be run from a pre-built Docker image that has everything needed already installed. This should make the setup much
easier for users as well as reducing the need to install lots of software on their local machines. Running via Docker is obviously less flexible in terms of the configuration of the
third-party software but has been built with the most common use cases in mind and reducing the image size to as small as possible, without losing any of requirements oligo uses from
the third-party software. Currently the Docker image only supports running oligo with BLAT, not STAR.
First pull the latest oligo image onto your local machine:
$ docker pull jbkerry/oligo:latest
You can also specify a version if needed. The Docker image versions match the oligo package version i.e., jbkerry/oligo:0.2 will be running oligo v0.2:
$ docker pull jbkerry/oligo:0.2
The docker entrypoint is set to run oligo with the config file already set up to point to the install executables of BLAT and RepeatMasker so users can run the image, starting with
the oligo subcommand that is required (see the Usage section for more details).
In order for your BED file and reference genome FASTA files to be accessible to the Docker container, your local directories with these files must be mounted into the Docker container
using the -v option when you call the docker run command on the image. The Docker image runs the oligo command from a top-level directory called /results and stores
all of its output files here. In order to see them on your local machine after the run has finished, you will need to mount a local directory where you want to store the results, to
this /results directory. Again, this mount with the -v option needs to be done at the image runtime.
For example, running the oligo command with the off-target subcommand might look something like this with the Docker image:
$ docker run -v /local/path/oligo_results:/results -v /local/human:/genome jbkerry/oligo:latest off-target -f /genome/genome.fa -g hg38 -b ./off_target_sites.bed -o 100 -t 50 -m 300 --blat
With this command, the output results will appear in the example local directory of /local/path/oligo_results. Note that this example command is using the Linux filepath
format (i.e., /.../) for the local directories. On Windows (not using WSL) the mounting would look like this:
$ docker run -v C:\local\path\oligo_results:/results -v C:\local\human:/genome jbkerry/oligo:latest off-target -f /genome/genome.fa -g hg38 -b ./off_target_sites.bed -o 100 -t 50 -m 300 --blat
Because the docker image is built on top of a Debian Linux image, the paths that local directories get mounted to in the container (i.e. the right-hand side of the : for
the -v options) still need to use the Linux filepath format, even when running from a Windows machine.
Installation specifics¶
- Below is a list of the versions and alterations that have been made to the standard installs of third-party software for the
oligoDocker image: RepeatMasker v4.1.5
Dfam.h5 library has been replaced with an HMM matrices containing only mouse- and human-specific transposable elements in order to reduce the size of the Docker image
HMMER v3.3.2
Tandem Repeat Finder v4.09.1
BLAT v37.x1
The HMM matrices were generated with the following two commands, run from with the top-level RepeatMasker directory (famdb.py comes bundled with the latest versions of RepeatMasker):
$ ./famdb.py -i Libraries/RepeatMaskerLib.h5 families --format hmm 'Homo sapiens' --include-class-in-name >humans.hmm
$ ./famdb.py -i Libraries/RepeatMaskerLib.h5 families --format hmm 'Mus musculus' --include-class-in-name >mouse.hmm
The Dockerfile in the oligo GitHub repository can be referenced for details of the how the Docker image was built. Some reference data files that get copied into the image at build
time are not present in the repository but can be provided to the user if needed.
Usage¶
oligo can be run with one of three subcommands
capture: designs oligos for a standard Capture-C experiment. The user supplies a list of viewpoint coordinates, and oligos are generated adjacent to the flanking recognition sequence of a specified restriction enzyme.
tiled: designs oligos for multiple adjacent restriction fragments across a specified region of a chromosome, or for the entire chromosome. If
tiledis run in contiguous mode, oligos are generated independent of restriction fragments and are instead generated for a user-specified step size, in an adjacent manner.off-target: designs oligos to capture DNA surrounding potential CRISPR off-target cut sites to allow for efficient sequencing to determine off-target activity.
These subcommands all generate oligo sequences, based on different underlying behaviours. Methods from the Tools class in the oligo.tools module are then used to check
the off-target binding and repeat content of the oligos. This information is output in a file called oligo_info.txt; oligo sequences are written to a FASTA file called oligo_seqs.fa
Example
The subcommand follows the oligo command and options for the subcommand are then specified afterwards. Note that the config text file, specifying paths to the installed RepeatMasker,
BLAT and STAR directories (see Dependencies) must be specified between oligo and the chosen subcommand, with the -cfg argument. An example config file can be viewed
here. Below, is an example using the off-target subcommand:
$ python -m oligo -cfg ./config.txt off-target -f /path/to/human/genome.fa -g hg38 -b ./off_target_sites.bed -o 100 -t 50 -m 300 --blat
Command-line help
The oligo options and subcommands can be viewed with the --help flag from the command-line:
$ python -m oligo --help
Usage: python -m oligo [OPTIONS] COMMAND [ARGS]...
Options:
--version
-cfg, --config PATH [required]
--help Show this message and exit.
Commands:
capture
off-target
tiled
For options specific to each subcommand, run oligo with the desired subcommand, followed by --help. Note that you will need to provide the -cfg flag for this to work:
$ python -m oligo -cfg ./config.txt off-target --help
Usage: python -m oligo off-target [OPTIONS]
Options:
-f, --fasta PATH Path to reference genome fasta. [required]
(etc.)
More detailed usage information for each of the subcommads can be found in the individual pages, via the navigation on the left.
Pipeline Schematic¶
A schematic of the pipeline workflows is shown below. The tools.Tools class is where the RepeatMasker and BLAT or STAR software is run to determine the off-target alignments as as the simple sequence repeats of each generated oligo.
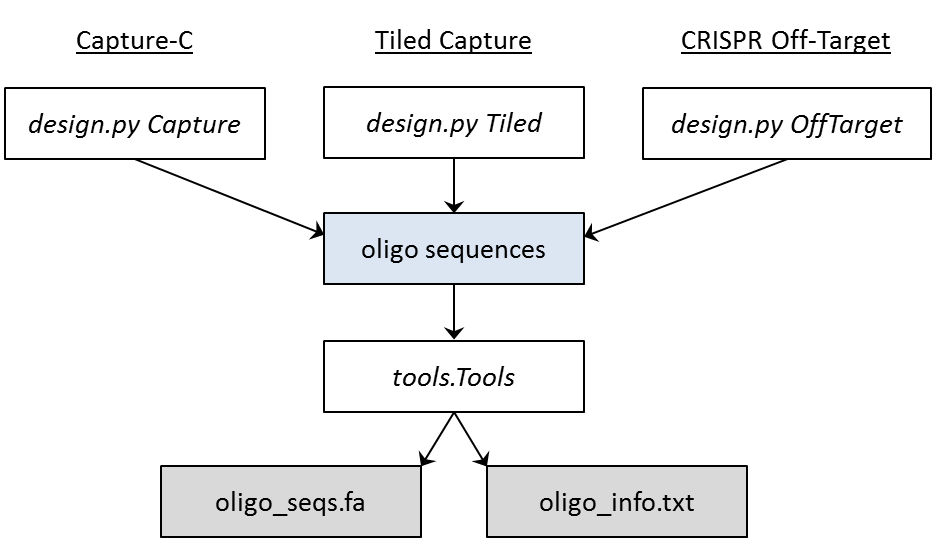
Workflows of the three pipelines¶
Output¶
Although a number of files, including alignment files, are output from the pipelines, the important one is oligo_info.txt. This is a tab-delimited text file that contains the following information for every oligo:
- chr
the chromsome that the oligo/fragment is on
- start
the bp start coordinate of the oligo
- stop
the bp stop coordinate of the oligo
- fragment_start
the bp start coordinate of the fragment
- fragment_stop
the bp stop coordinate of the fragment
- side_of_fragment
the side of the fragment that the oligo is situated on (left or right)
- sequence
the full DNA sequence of the oligo
- total_number_of_alignments
the total number of times the oligo was found in the BLAT/STAR alignment file
- density_score
the base-pair average and length-normalised number of times the oligo was found in the BLAT/STAR alignment file (see Choosing Good Oligos for more details)
- repeat_length
the length of the longest simple sequence repeat found in the oligo
- repeat_class
the class of the longest simple sequence repeat found in the oligo
- GC%
the GC percentage of the oligo sequence
- associations
the position/gene name associated with this oligo; this is the viewpoint name supplied in the bed file (4th column)
Missing Values¶
Due to particular differences between the three pipelines, and in order to keep a consistent output format between the three, there are instances where some values in the file will be purposefully missing.
- fragment_start, fragment_stop, side_of_fragment
These values will be replaced with a ‘.’ for the Tiled Capture pipeline when run in contiguous mode, and for the CRISPR Off-Target pipeline, as both of these pipelines are restriction fragment-independent
- associations
This value will be replaced with a ‘.’ for the Tiled Capture pipeline as these oligos are generated for adjacent sites across one large region and not for different viewpoints associated with unique names
Choosing Good Oligos¶
Note
Cut-offs for efficient oligos:
Density Score¶
When performing a Capture experiment it is important that the oligo is not susceptible to off-target binding as this would result in the pulldown of unwanted material, generating false positives. To assess the degree of off-target binding, each oligo is aligned against the genome using either STAR or BLAT (for information on choosing between STAR or BLAT see any of the individual pipeline documentation). The number of times an oligo aligns to the genome is used to assess its degree of off-target binding and this value is represented by the density score. Density score is calculated by counting the base-pair coverage of each alignment for an oligo and dividing that value by the length of the oligo. A schematic of this calculation is shown below, for two 70bp oligos.
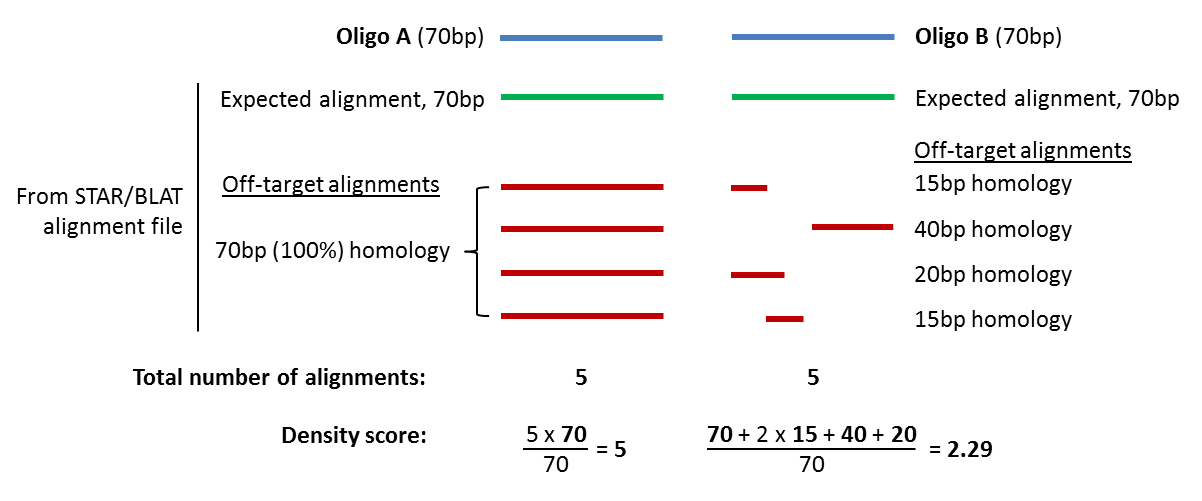
Therefore, a lower density score is better, with the exception of values below 1. A value between 0 and 1 means no perfect alignment was found so the pulldown will not be 100% efficient. A score of 0 means that either no alignments were found so this oligo will not capture anything or, in the case of using STAR, can also mean that it aligned so many times that it passed the threshold for a read to be ignored, and hence would have a high degree of off-target binding. A safe cut-off for density score, to ensure efficient oligo capture without problematic off-target binding, is greater than or equal to 1 and less than or equal to 30 (if using BLAT), or greater than or equal to 1 and less than or equal to 50 (if using STAR).
Repeat Length¶
The presence of simple sequence repeats in an oligo can also cause a greater degree of off-target binding due to the ambiguous presence of these repeats throughout the genome. Usually if an oligo has a long repeat within it, it will also have a high density score. However, it is still best to filter on repeat length as well. We filter for oligos that have a repeat length less than or equal to a quarter of the length of the oligo, so for an 80bp oligo only those with a repeat length less than or equal to 20 would be accepted. Unlike density score, 0 is the best value as this means that the oligo does not contain any simple sequence repeats.